“The Impact of Autophagy on Pancreatic Ductal Adenocarcinoma (PDAC) Development” won first place for the 2017 S. Klein Prize for Technical Writing.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer death and lacks effective treatment. Although much of the mechanisms underlying PDAC development is still unknown, autophagy – the process of catabolizing damaged cellular components and bioenergetic macromolecules – has been found to impact PDAC growth. Although some studies have found that autophagy inhibition drives PDAC progression, inhibition of autophagy via gene deletions or drug administration has generally been found to prevent PDAC progression. Through these studies, several genes associated with autophagy have been found to be particularly important in PDAC development, including the autophagy-related (ATG) genes, SQSTM1, CYB5A, and possibly TP53. Although there have been attempts to target these specific genes as a means of PDAC therapy, a more promising form of treatment for PDAC currently appears to be the use of autophagy inhibitors, such as chloroquine or hydroxychloroquine, in conjunction with chemotherapeutics such as gemcitabine. This review will focus on the role that autophagy plays in PDAC development, the essential genes that control autophagy, and targeting autophagy as a means of PDAC therapy.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer, is a malignancy associated with poor prognosis and expected to become the second most common cause of cancer death by 2030.1 PDAC originates in the exocrine ducts of the pancreas that are responsible for secreting enzymes that digest carbohydrates, proteins, and lipids. Because early stages of PDAC do not elicit clear symptoms, patients are often not diagnosed until after the tumor has metastasized to other parts of the body. This difficulty in early diagnosis gives PDAC a 5-year survival rate of less than 5%.
Although combined treatment with gemcitabine (Table 1, Therapy 1) and albumin-bound paclitaxel (Table 1, Therapy 2) has been found to be a promising treatment for PDAC2 along with the ONIVYDE® (irinotecan liposome injection)3 (Table 1, Therapy 3), there is still a general lack of available therapy for the disease because not enough detail is known about the mechanisms of PDAC development.
Recent studies have, however, discovered that autophagy may be significantly involved in PDAC development. With multiple discoveries that PDAC is driven by KRAS (Table 1, Gene 1) mutations4,5,6, and from recent evidence suggesting that autophagy is required for KRAS-mutated tumors to survive7,8, it has become probable that autophagy is necessary for PDAC development. Researchers have therefore begun to focus specifically on the link between PDAC development and autophagy.
Autophagy is the natural cellular process of degrading macromolecules and organelles into lipids, amino acids, and nucleosides for reuse. It maintains cellular homeostasis by ridding cells of damaged components and catabolizing macromolecules for energy. This breakdown of damaged components and bioenergetic macromolecules prevents organelle or protein malfunction and allows self-nourishment in response to starvation, making autophagy particularly important at early neonatal stages when amino acids are especially lacking.9
Autophagy thus has a straightforward function, but it can occur through different pathways. These pathways are known as macroautophagy, microautophagy, and chaperone-mediated autophagy. This review will focus on macroautophagy because it is the most common pathway.
In macroautophagy, a double-membraned, spherical structure known as an autophagosome forms around the target cellular component and then fuses with a lysosome to degrade the internalized target. Although this is a simple mechanism, its underlying molecular pathway is complex10 (Cheong, et al. (2012), Figure 1). The induction of autophagy begins with the formation of a phagophore, a double-membraned structure, via protein complexes involving kinases and autophagy-related (ATG) genes (Table 1, Gene 2). The phagophore then elongates to form an autophagosome through two ubiquitin-like conjugation systems involving additional ATG genes and LC3, a protein that interacts with microtubules. The completed autophagosome then fuses with a lysosome to degrade internalized cellular components and to conclude the process of autophagy.
Although the autophagy pathway is thus well mapped, it was only recently discovered that this pathway plays a crucial role in PDAC development. Through disrupting genes in the autophagy pathway and observing their effects on tumor development, researchers have found that autophagy helps control PDAC progression.11,12 Because of this finding, current research has begun focusing on targeting autophagy to potentially treat PDAC. This review will therefore explore autophagy’s impact on PDAC development while highlighting the genes involved and the current findings on autophagy-related PDAC treatment.
The role of autophagy in PDAC progression
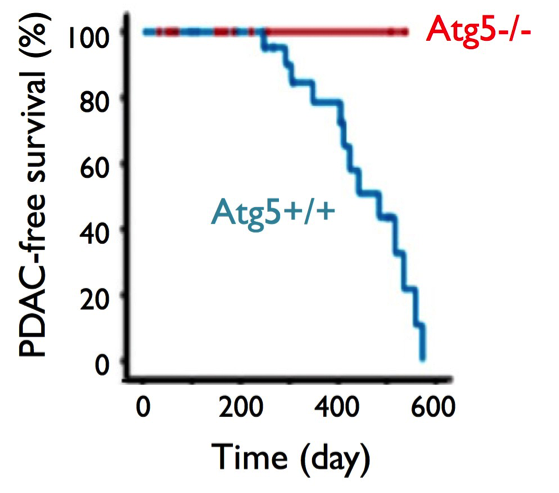
Although whole-exome sequencing studies have identified several mutations characteristic of PDAC cells,13,14 additional genes – particularly those involved in autophagy – have recently been found to influence the progression of PDAC. For example, one study has shown that the deletion of ATG genes (Table 1, Gene 2) blocks the development of PDAC into a high-grade, or late-stage, tumor, despite the formation of pre-malignant intraepithelial neoplasias (PanIN), which are pre-cancerous abnormal growths of epithelial tissue15 (Rosenfeldt, et al. (2013), Figure 2). Similarly, a study by Yang, et al. has demonstrated that the inhibition of ATG5 with siRNA significantly prevents PDAC cell proliferation16 (Yang, et al. (2011), Figure 3). Finally, PDAC tissues have been found to have significant levels of ATG gene expression.17 Together, these findings showcase the necessity for ATG genes and autophagy in PDAC growth.
In addition, some studies have verified the need for autophagy in PDAC development by treating PDAC cells with autophagy-inhibitory drugs, rather than by mutating specific genes in the autophagy pathway. For example, Yang, et al. treated PDAC tumors with chloroquine (Table 1, Therapy 4) to prevent autophagosome degradation and showed that autophagy inhibition prevented further tumor growth.18
Tumor growth may have been prevented because cancer cells possibly rely heavily on autophagy to recycle proteins and generate the energy required for continuous proliferation. Without this regenerative process, PDAC tumors cannot grow. Because the inhibition of autophagy thus prevents PDAC progression, PDAC cells most likely require autophagy activation for survival and proliferation.
However, despite the evidence that supports the need for autophagy activation in PDAC growth, some studies have found that autophagy inhibition, rather than activation, promotes tumor progression in other organs. For example, Mathew, et al. found that lung cells with defective autophagy intracellularly accumulate endoplasmic reticulum (ER) chaperones, reactive oxygen species (ROS), and p62 – a protein that helps target proteins into autophagosomes for degradation – and that this accumulation causes tumor growth.19 These accumulations could cause stress and DNA damage responses, which induce cell division abnormalities and genetic instabilities that promote tumor growth. In addition, Takamura, et al. have found that autophagy-deficient mouse models with an ATG5 and liver-specific ATG7 deletion exhibit liver tumorigenesis due to p62 accumulation and stress.20 Although these studies are not directly related to PDAC, because they showed that ATG deletion could cause tumor growth in general, there still might be a possibility that autophagy inhibition induces growth in other cancers like PDAC.
A possible explanation for this apparent dual role of autophagy is that autophagy impacts PDAC development in a stage-specific manner. Researchers have hypothesized that PDAC requires autophagy inhibition early in development but requires autophagy activation later on.21 The theory states that on the one hand, autophagy inhibition causes p62, ER chaperones, and ROS to accumulate and promote cellular stress,22 which thereby creates an environment conducive to tumorigenesis. On the other hand, once the tumor has formed, autophagy activation is necessary to recycle damaged cellular components and allow the tumor to continue survival. Further research is needed, however, to clarify this theory.
Essential genes involved in autophagy and in PDAC
ATG3, ATG5, and ATG7
Autophagy dysregulation arises from mutations in genes involved in autophagosome formation. Several genes – including ATG3, ATG5, and ATG7 – control the formation of autophagosomes, and a recent study has found that mutations in ATG5 and ATG7 reduce tumor growth in KRAS-oncogenic (Table 1, Gene 1) PDAC cell lines.23 KRAS-expressing ATG5 or ATG7 knockout mice exhibited reduced tumor growth, suggesting that autophagy promotes KRAS-mediated tumor development (Guo, et al. (2011), Figure 4). Similarly, Lock et al. have found that ATG3, ATG5, or ATG7 deletions reduce cell growth in KRAS-mutated cell strains.24 Because most PDAC cells are KRAS-mutated, this finding suggests that the deletion of ATG genes, and therefore the inhibition of autophagy, diminishes PDAC proliferation.
SQSTM1
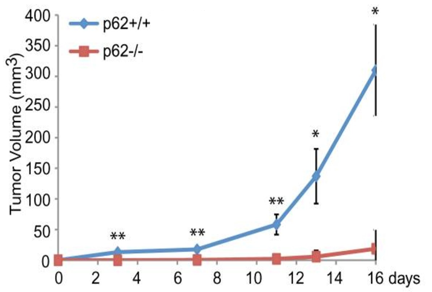
In addition to the ATG genes, the SQSTM1 gene, which encodes for the p62 protein, appears to affect PDAC progression through its role in autophagy. P62 is a nucleoporin and autophagy cargo receptor that binds ubiquitin on modified proteins and targets them to LC3 on autophagosomes. Through its mediatory role as a cargo receptor, p62 is needed for autophagy and hence for efficient PDAC growth. This relationship between p62 and PDAC was shown by the lack of PDAC growth in p62 knockouts.25 In addition, KRAS-mutated p62 knockout cells have been shown to reduce tumor growth (Guo, et al. (2011), Figure 5), further suggesting that p62 is required for the development of KRAS-mutated tumors like PDAC.
MAPK and NF-κB pathways
Researchers have also begun studying external pathways that may indirectly activate autophagy. Some studies have decided to focus on the MAPK (Table 1, Pathway 1) and NF-êB (Table 1, Pathway 2) pathways because they include KRAS (Table 1, Gene 1), an oncogene in which most PDAC tumors are mutated. Because most PDACs have KRAS gain-of-function mutations and because PDACs require autophagy activation for growth, studies have hypothesized that the MAPK and NF-êB pathways indirectly activate autophagy for PDAC to continuously proliferate. For example, Papademetrio, et al. have discovered that the inhibition of the MAPK and NF-êB pathways using the U0126 (Table 1, Therapy 5) and caffeic acid phenethyl ester (CAPE) (Table 1, Therapy 6) drugs, respectively, halts autophagy and consequently induces PDAC cell apoptosis.26
This finding not only indicates that the MAPK and NF-êB pathways indirectly activate autophagy but also confirms that PDAC cells need autophagy for survival.
MALAT1
Besides the MAPK and NF-êB cascades, pathways associated with metastasis were also found to indirectly activate autophagy and influence PDAC growth. In a particular study, the elevated expression of MALAT1, a long noncoding RNA that regulates metastasis-associated protein expression, was found to increase autophagy and promote PDAC growth.27 A lack of MALAT1 appeared to inhibit autophagy activation by suppressing HuR, which is an mRNA regulator responsible for autophagy upregulation. In addition, the silencing of MALAT1 seemed to inhibit autophagy by enhancing regulation of TIA-1, an mRNA regulator that downregulates autophagy. Thus, genes, such as MALAT1, in external pathways appear to control autophagy and, consequently, PDAC progression.
TP53
Recent studies have also demonstrated that p53, a tumor suppressor protein encoded by the TP53 gene and located outside the autophagy pathway, may greatly influence autophagy’s role in PDAC growth. Specifically, the functional state of p53 – whether it is mutated or not – may determine whether autophagy promotes or inhibits PDAC growth. To illustrate, in a recent study by Rosenfeldt, et al., mice with p53- deficiency, PDAC, and mutated ATG7 showed accelerated tumor growth, while mice with p53-proficiency, PDAC, and mutated ATG7 did not. This finding implies that non- functional p53 diminishes the inhibitory effect that defective autophagy has on PDAC progression.28 Similar results were seen with mutated ATG5.
However, the validity of these results are questionable because Yang, et al., contrary to Rosenfeldt, et al., found that ATG5 deletions prevented PDAC progression in both p53-deficient and p53-proficient cells and that the state of p53 therefore did not matter.29 To account for this ambiguity, Yang, et al. concluded that this discrepancy arose due to differences in mouse models used. While Yang, et al. used mice in which p53 was lost by stochastic loss of heterozygosity (LOH), Rosenfeldt, et al. used mice that had p53 homozygously deleted during embryogenesis.30 Because human PDAC tumors lose p53 through stochastic LOH rather than through embryogenesis, however, Yang, et al. concluded that the Rosenfeldt group’s finding on p53 to be insignificant. Nevertheless, further research in this field is necessary to clarify if mutations in p53 influence PDAC development.
CYB5A
Similar to the way p53 possibly promotes PDAC growth when mutated, one final gene, which has recently been found to indirectly control autophagy, appears to influence PDAC growth in unexpected ways. This gene is known as CYB5A, which encodes the electron carrier cytochrome b5 that is located on cell membranes. CYB5A overexpression results in an increase in autolysosomes, suggesting that the expression of this gene upregulates autophagy. However, unlike the increase in PDAC growth associated with ATG expression, CYB5A expression appears to diminish this growth31] (Giovannetti, et al. (2014), Figure 6). This information therefore indicates that CYB5A, and therefore autophagy, may suppress PDAC tumor growth.
Targeting autophagy for PDAC therapy
Despite its ambiguous role in PDAC development, autophagy is a promising potential target for cancer treatment. Because PDAC cells respond to therapies, such as chemotherapy and radiation, by activating autophagy as a damage response, treating PDAC cells with both autophagy-inducing treatments and autophagy inhibitors could efficiently kill the PDAC cells. Scientists have therefore begun treating PDAC cells with both chemotherapeutic drugs and autophagy inhibitors to halt PDAC growth.
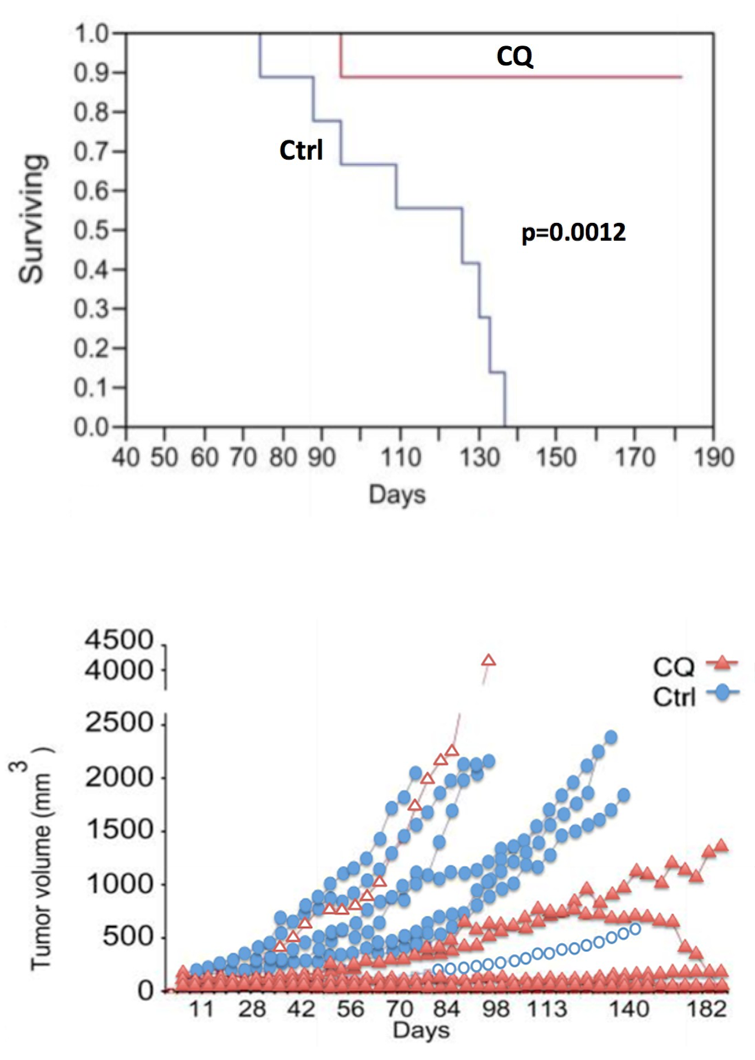
A common drug used to inhibit autophagy is chloroquine (Table 1, Therapy 4). Chloroquine prevents lysosomal acidification by antagonizing the effect of vacuole membrane protein 1 (VMP1)32; when lysosomal acidification is halted, lysosomal enzymes cannot function, autophagosome-lysosome fusion is obstructed, and autophagy is thus prevented. This autophagy inhibitor is an especially promising form of PDAC treatment because it was found to target PDAC tumor cells specifically in vitro33 and because it restricted tumor growth and prolonged survival of mice with PDAC xenografts (Yang, et al. (2011), Figure 7).34 Combined treatment of PDAC with chloroquine and gemcitabine (Table 1, Therapy 1) has been found to also significantly reduce tumor growth and prolong survival of mice with PDAC xenografts35 (Balic, et al. (2014), Figure 8), making these drugs a promising treatment. However, chloroquine is also unfortunately known to cause retinal toxicity, making it an unideal drug.
Recently, however, a derivative of chloroquine, known as hydroxychloroquine, has been studied as a more favorable treatment because of its lack of retinal toxicity; because hydroxychloroquine cannot cross the blood-retinal barrier, treated patients experience milder side effects. In their phase I/II clinical trial, Boone, et al. found that PDAC treatment with hydroxychloroquine and gemcitabine (Table 1, Therapy 1) effectively reduced tumor growth (50% of PDAC patients showed an over 50% reduction in CA 19-9, a PDAC marker),36 relative to treatment with gemcitabine alone (25% of PDAC patients showed an over 50% reduction in CA 19-9).37 Although hydroxychloroquine can also be administered without gemcitabine, a recent phase II clinical trial indicated that treatment of PDAC with hydroxychloroquine alone showed negligible and inconsistent efficacy.38 Hydroxychloroquine is therefore a promising treatment for PDAC when used in conjunction with gemcitabine.
Analogous to the way in which hydroxychloroquine works effectively with gemcitabine, another drug named verteporfin appears to halt PDAC growth when treated together with gemcitabine. Verteporfin, which inhibits autophagosome formation,39 is extremely toxic to PDAC cells, but it alone cannot reduce tumor growth; it only has the power to interrupt tumor growth by preventing autophagy. However, it has been discovered that co-treatment of verteporfin with gemcitabine reduces tumor growth and promotes longer survival.40 This finding suggests that verteporfin would be an effective treatment for PDAC when combined with gemcitabine. In addition, phase I/II clinical trials with verteporfin have confirmed the drug’s safety and lack of adverse effects.41
Therefore, autophagy inhibition via verteporfin seems to effectively reduce PDAC tumor progression.
Although such use of an autophagy inhibitor in conjunction with chemotherapy shows promising effects as a therapeutic, lumican, an extracellular matrix proteoglycan overexpressed in PDAC cells,42 may be enhancing this therapeutic effect in PDACs.
When Li, et al. treated PDAC cells with gemcitabine (Table 1, Therapy 1), lumican secretion increased and autophagosome formation decreased significantly in response.43 Lumican thus appears to mimic the role of autophagy inhibitors when exposed to gemcitabine. Therefore, it is unclear whether the lumican or the autophagy inhibitors are reducing tumor growth in clinical trials that test the efficacy of combined treatment with autophagy inhibitors and gemcitabine. Although the mechanism by which lumican helps inhibit PDAC growth is unclear, Li, et al. have suggested that lumican triggers mitochondrial malfunction and induces sensitivity to gemcitabine. Further study is necessary, however, to determine whether or not lumican is enhancing the effects of autophagy inhibitors and chemotherapy.
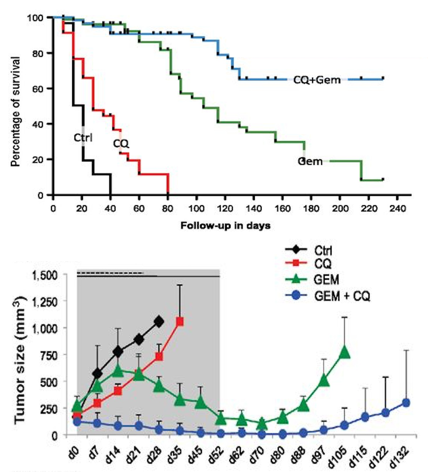
It has additionally been found that the co-treatment of MAPK (Table 1, Pathway 1) or NF-êB (Table 1, Pathway 2) pathway inhibitors along with autophagy inhibitors may impede PDAC growth. In the Papademetrio study, PDACs treated with U0126 (Table 1, Therapy 5) or CAPE (Table 1, Therapy 6) did not grow further.44 However, PDAC growth was halted without inducing apoptosis; the tumor cells, therefore, had not been killed. Apoptosis was only induced if autophagy was inhibited.45 Therefore, this combined treatment with MAPK or NF-êB inhibitors and autophagy inhibitors is a promising therapy for PDAC, and it is now necessary to run further clinical trials on this drug combination.
Finally, the ATG genes (Table 1, Gene 2) could possibly be targeted directly for PDAC treatment. For example, as Ko, et al. have demonstrated, deletions of genes such as ATG5 could offer potential therapeutic treatments for PDAC because human PDAC patients with higher protein expression levels of ATG5 tended to have shorter survival than those with lower levels of ATG5.46 No such drugs have been developed yet, however, and further study is needed for its generation.
Conclusion
Autophagy plays a vital role in PDAC progression through its destruction of damaged cellular components and catabolism of bioenergetics macromolecules. Such catabolic properties are especially necessary for tumor cells to survive in the hypoxic and nutrient-deficient environment. Although some evidence suggests that autophagy prevents PDAC growth, most studies indicate that it promotes PDAC progression. In fact, not only do genes directly related to autophagy – such as ATG5 and ATG7 – contribute to PDAC development, but genes of indirectly-related pathways – such as SQSTM1 or CYB5A – appear to contribute to PDAC growth as well. Because autophagy thus appears to be crucial to PDAC survival, clinical trials with autophagy inhibitors are under progress, including trials with hydroxychloroquine and verteporfin. These autophagy inhibitors seem to be especially effective when administered in conjunction with chemotherapeutic drugs such as gemcitabine. In addition, inhibitors of outside pathways that are indirectly related to autophagy – such as the MAPK or NF-êB pathways – also seem to successfully reduce PDAC growth, providing a wider range of possible sources for therapeutic drugs against PDAC. However, because autophagy has thus been found to have both pro-tumorigenic and anti-tumorigenic effects, and because hydroxychloroquine has been found to lack efficacy alone, it is necessary to focus future studies on clarifying autophagy’s role in tumorigenesis and illuminating the true effectiveness of certain therepeutics. By continuing studies on current treatments while searching for new autophagy inhibitors as well, it is possible to accelerate the improvement of PDAC treatment methods.
Tables and Figures
| Gene | Protein Function |
| 1. KRAS | An oncogenic GTPase involved in signal transduction pathways of cell division and differentiation47 |
| 2. Autophagy-related (ATG) genes | Genes involved in autophagy activation48 |
| Pathway Function | |
| 1. MAPK | A pathway involving kinases that controls cell proliferation, gene expression, differentiation, and apoptosis49 |
| 2. NF-ĸB | A pathway that controls transcription of genes involved in innate and adaptive immunity50 |
| Therapy Definition | |
| 1. Gemcitabine | A chemotherapeutic agent that acts as a nucleoside analog and disrupts DNA replication in cells51 |
| 2. Paclitaxel | A chemotherapeutic agent that prevents normal microtubule disintegration52 |
| 3. ONIVYDE® (irinotecan liposome injection) | A treatment that delivers irinotecan, a chemotherapy drug, efficiently to tumor sites by encapsulating irinotecan in liposome nanoparticles (produced by Merrimack Pharmaceuticals, Inc.)53 |
| 4. Chloroquine | An autophagy inhibitor that prevents lysosomal acidification54 |
| 5. U0126 | A selective inhibitor of the MEK1 and MEK2 genes in the MAPK pathway55 |
| 6. Caffeic acid phenethyl ester (CAPE) | An inhibitor of the NF-κB pathway56 |
Figures




References
1 Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Research. 2014;74:2913–2921. ⬏
2 Von Hoff DD, Ervin T, Arena FP, Chiorean EG, et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. New England Journal of Medicine. 2013;369:1691–1703. ⬏
3 Wang-Gillam A, Li CP, Bodoky G, Dean A, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine- based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. 2016;387:545-57. ⬏
4 Witkiewicz AK, McMillan EA, Balaji U, Baek G, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature Communications. 2015;6:6744. ⬏
5 Sausen M, Phallen J, Adleff V, Jones S, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nature Communications. 2015;6:7686. ⬏
6 Collins MA, Bednar F, Zhang Y, Brisset JC, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. Journal of Clinical Investigation. 2012;122:639–653. ⬏
7 Kim MJ, Woo SJ, Yoon CH, Lee JS, et al. Involvement of autophagy in oncogenic K- Ras-induced malignant cell transformation. Journal of Biological Chemistry. 2011;286:12924-32. ⬏
8 Guo JY, Chen HY, Mathew R, Fan J, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes & Development. 2011;25:460–470. ⬏
9 Kuma A, Hatano M, Matsui M, Yamamoto A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. ⬏
10 Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nature Biotechnology. 2012;30:671-678. ⬏
11 Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. ⬏
12 Guo, et al. ⬏
13 Murphy SJ, Hart SN, Lima JF, Kipp BR, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology. 2013;145:1098-1109. ⬏
14 Xie T, Musteanu M, Lopez-Casas PP, Shields DJ, et al. Whole exome sequencing of rapid autopsy tumors and xenograft models reveals possible driver mutations underlying tumor progression. PLoS One. 2015;10:e0142631. ⬏
15 Rosenfeldt, et al. ⬏
16 Yang S, Wang X, Contino G, Liesa M, et al. Pancreatic cancers require autophagy for tumor growth. Genes & Development. 2011;25:717–729. ⬏
17 Ko YH, Cho Y-S, Won HS, Jeon EK, et al. Prognostic significance of autophagy- related protein expression in resected pancreatic ductal adenocarcinoma. Pancreas. 2013;42:829–35. ⬏
18 Yang, et al. (2011). ⬏
19 Mathew R, Karp CM, Beaudoin B, Vuong N, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. ⬏
20 Takamura A, Komatsu M, Hara T, Sakamoto A, et al. Autophagy-deficient mice develop multiple liver tumors. Genes & Development. 2011;25:795–800. ⬏
21 Yang A, Kimmelman AC. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy. 2014;10:1683-84. ⬏
22 Mathew, et al. ⬏
23 Guo, et al. ⬏
24 Lock R, Roy S, Kenific CM, Su JS, et al. Autophagy facilitates glycolysis during Ras- mediated oncogenic transformation. Molecular Biology of the Cell. 2011;22:165–178. ⬏
25 Guo, et al. ⬏
26 Papademetrio DL, Lompardía SL, Simunovich T, Constantino S, et al. Inhibition of survival pathways MAPK and NF-kB triggers apoptosis in pancreatic ductal adenocarcinoma cells via suppression of autophagy. Targeted Oncology. 2016;11:183-95. ⬏
27 Li L, Chen H, Gao Y, Wang YW, et al. Long noncoding RNA MALAT1 promotes aggressive pancreatic cancer proliferation and metastasis via the stimulation of autophagy. Molecular Cancer Therapeutics. 2016;15:2232-43. ⬏
28 Rosenfeldt, et al. ⬏
29 Yang, et al. (2014). ⬏
30 Yang, et al. (2014). ⬏
31 Giovannetti E, Wang Q, Avan A, Funel N, et al. Role of CYB5A in pancreatic cancer prognosis and autophagy modulation. Journal of the National Cancer Institute. 2014;106:djt346. ⬏
32 Loncle C, Molejon MI, Lac S, Tellechea JI, et al. The pancreatitis-associated protein VMP1, a key regulator of inducible autophagy, promotes KrasG12D-mediated pancreatic cancer initiation. Cell Death & Disease. 2016;7:e2295. ⬏
33 Zeilhofer H, Mollenhauer J, Brune K. Selective growth inhibition of ductal pancreatic adenocarcinoma cells by the lysosomotropic agent chloroquine. Cancer Letters. 1989;44:61–66. ⬏
34 Yang, et al. (2011). ⬏
35 Balic A, Sorensen MD, Trabulo SM, Sainz B Jr, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Molecular Cancer Therapeutics. 2014;13:1758-71. ⬏
36 Boone BA, Bahary N, Zureikat AH, Moser AJ, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine patients with pancreatic adenocarcinoma. Annals of Surgical Oncology. 2015;22:4402-10. ⬏
37 Hammand N, Heilburn LK, Philip PA, Shields AF, et al. CA19-9 as a predictor of tumor response and survival in patients with advanced pancreatic cancer treated with gemcitabine based chemotherapy. Asia-Pacific Journal of Clinical Oncology. 2010;6:98– 105. ⬏
38 Wolpin, BM, Rubinson DA, Wang X, Chan JA, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. The Oncologist. 2014;19:637–638. ⬏
39 Donohue E, Thomas A, Maurer N, Manisali I, et al. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. Journal of Cancer. 2013;4:585-596. ⬏
40 Donohue, et al. ⬏
41 Huggett MT, Jermyn M, Gillams A, Illing R, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. British Journal of Cancer. 2014;110:1698–1704. ⬏
42 Li X, Roife D, Kang Y, Dai B, et al. Extracellular lumican augments cytotoxicity of chemotherapy in pancreatic ductal adenocarcinoma cells via autophagy inhibition. Oncogene. 2016:15;35:4881-90. ⬏
43 Li, et al. ⬏
44 Papademetrio, et al. ⬏
45 Papademetrio, et al. ⬏
46 Ko, et al. ⬏
47 O’Leary NA, Wright MW, Brister JR, Ciufo S, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733-45. ⬏
48 O’Leary, et al. 49 O’Leary, et al. 50 O’Leary, et al. 51 O’Leary, et al. 52 O’Leary, et al. 53 O’Leary, et al. 54 O’Leary, et al. 55 O’Leary, et al. 56 O’Leary, et al. ⬏
49 O’Leary, et al. ⬏
50 O’Leary, et al. ⬏
51 O’Leary, et al. ⬏
52 O’Leary, et al. ⬏
53 O’Leary, et al. ⬏
54 O’Leary, et al. ⬏
55 O’Leary, et al. ⬏
56 O’Leary, et al. ⬏



